
JOURNAL OF CHEMICAL INFORMATION AND MODELING
Protein pocket matching, or binding site comparison, is of importance in drug discovery. Identification of similar binding pockets can help guide efforts for hit finding, understanding polypharmacology and characterization of protein function. The design of pocket matching methods has traditionally involved much intuition, and has employed a broad variety of algorithms and representations of the input protein structures. We regard the high heterogeneity of past work and the recent availability of large-scale benchmarks as an indicator that a data-driven approach may provide a new perspective. We propose DeeplyTough, a convolutional neural network that encodes a three-dimensional representation of protein pockets into descriptor vectors that may be compared efficiently in an alignment-free manner by computing pairwise Euclidean distances.
Proteins in small molecule drug discovery
Proteins perform roles in the body and are responsible for biological function. In drug discovery, we often seek to change a particular protein’s function to help the body combat disease. Drug discovery efforts that focus on modulating a particular protein with a small molecule drug can be framed as target-based drug discovery (where the protein is the target). A protein’s biological function is determined by its structure and sequence (proteins are constructed from chains of amino acids). The most common method for affecting a protein’s function is to interfere with a protein-ligand binding site, or pocket, capable of accommodating small molecules. A commonly used analogy is that the protein is a lock, and the drug is a key, and in drug discovery the game is to design the optimal key that fits in the lock. Obviously this is a simplification, not least because the key and lock are both constantly moving!
Similarity in target protein space
Figuring out which proteins a molecule may bind to is a task at the core of drug discovery. Depending on your start point, you may know a molecule binds one protein but suspect others to be interacting also, this can contribute to drug toxicity or polypharmacology. Conversely you may know of no molecules that bind your protein of interest, and are hoping to draw inspiration from molecules known to bind similar protein binding sites in a virtual screening scenario.
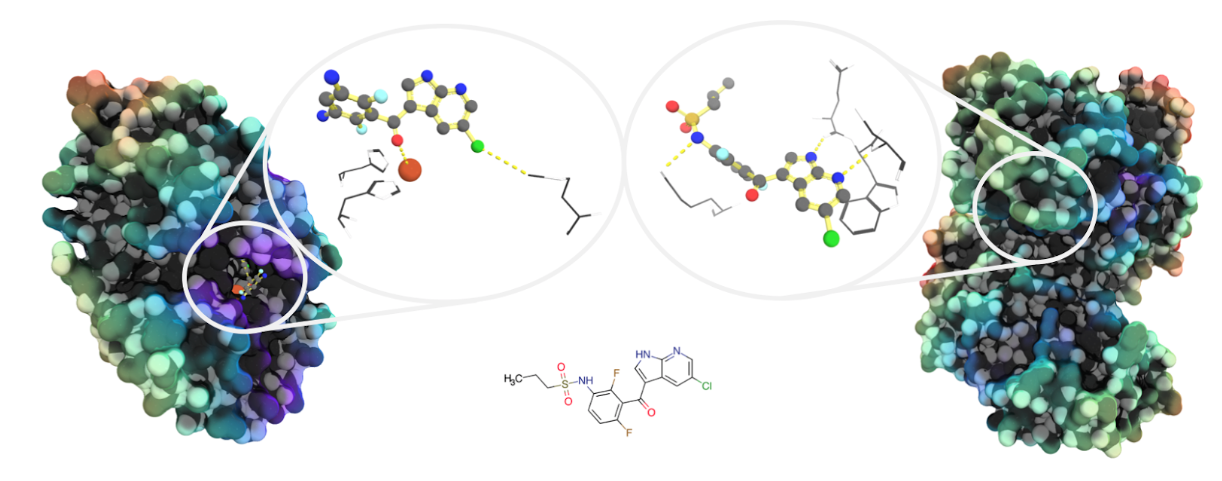
Image: Unrelated proteins may bind similar molecules. Both pirin (left, PDB ID: 6H1H) and the protein kinase B-Raf (right: PDB ID: 4W05) have been shown to bind analogues of the vemurafenib progenitor molecule PLX-4720
Traditional pocket matching approaches
Traditional approaches for assessing the similarity between 3D protein pockets are often based on a combination of hand crafted representations of the pockets and traditional shape matching algorithms. We hypothesised that a machine learning method for pocket matching would be able to learn a pocket representation directly from the data, while simultaneously learning which features of a pocket are particularly important for shared molecule binding. By introducing a machine learning-based solution to the problem of pocket matching we hoped to remove human bias and produce a pocket representation tailored for drug discovery.
Deeplytough: a machine learning method for 3d pocket matching
We view protein pocket matching through the lens of computer vision where we regard 3D protein structures as 3D images. Convolutional Neural Networks (CNNs) have proved effective across a number of domains and are able to build powerful representations of the input from low level pixels – except in 3D, pixels are known as voxels. In contrast to images, where each pixel is represented by intensities of red, green and blue (rgb), we replace the additive primary colours with molecular properties such as hydrophobicity or the ability of an atom to form hydrogen bonds.
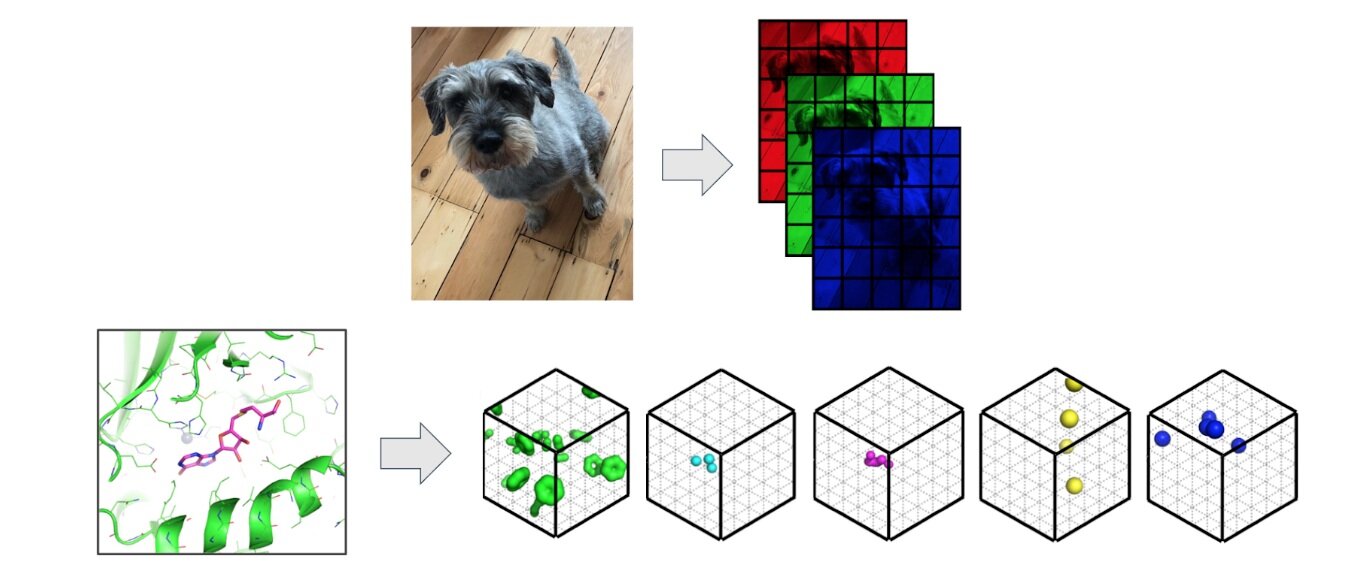
Image: Protein structures are featured as 3D images where pixels associated with intensities of red, green and blue are replaced by 3D voxels associated with descriptions of potential intermolecular interactions
To train DeeplyTough, we take advantage of a recently published dataset of ~1 million data points, TOUGH-M1.
Machine Learning
The field of distance metric learning aims to automatically determine the distance between items, and can be trained in a machine learning manner given a dataset of labelled examples. In our case, the TOUGH-M1 dataset contains examples of positive pairs (two protein pockets that have been shown to bind similar molecules), and negative pairs (two protein pockets that have shown no evidence of shared molecule binding). We train DeeplyTough to encourage separation of negative pairs, and close proximity of positive pairs in euclidean space.
We thoroughly benchmark our approach using three publicly available datasets and demonstrate that a metric learning approach for 3D pocket matching can show performance that is competitive with existing approaches on a much faster timescale at runtime. Our work represents a proof of concept showing that metric learning is capable of matching protein pockets that are likely to bind similar ligands directly from 3D protein structure data.An approach for protein pocket matching is one tool in the toolkit of modern drug discovery. We have introduced DeeplyTough, the first learned method for matching protein pockets, this shift in paradigm reduces the human bias associated with previous approaches, and reduces the runtime cost allowing us to consider much larger datasets. DeeplyTough will accelerate our ability to effectively search for novel drugs.
Back to publications